
Reacciones de adición y eliminación.
Las adiciones son usadas frecuentemente en síntesis para convertir un alqueno en otra función química. Las eliminaciones son, mecanísticamente, el proceso inverso y sirven para "formar" dobles uniones.Debido a las similitudes en el mecanismo y requerimientos estereoelectrónicos, los dos tipos de reacciones pueden ser estudiados en conjunto.
Algunos de los principales reactivos usados para adiciones a dobles enlaces son los siguientes:
H₂/cat ➖cis⟶ alcanos
BH₃/H₂O₂ ➖cis⟶ monoalcoholes
BH₃/AcOH ➖cis⟶ alcanos
OsO₄ ➖cis⟶ cisdioles
KMnO₄ ➖cis⟶ cisdioles
R-COOH ➖cis⟶ epóxidos ➖hidrólisis⟶ transdioles
Carbenos ➖cis⟶ ciclopropanos
Ozono ➖cis⟶ ozónido ➖hidrólisis⟶ aldehídos, cetonas, alcoholes o ácidos
Dienófilo + dieno ➖cis⟶ anillo de seis miembros
HX ➖trans⟶ monohalogenuros
H₂O/H⁺ ➖trans⟶ monoalcoholes
H₂O/X₂ ➖trans⟶ halohidrinas
X₂ ➖trans⟶ dihalogenuros

En este último caso se verifica además que son diaxiales. El electrófilo activo ataca por el lado menos impedido de la doble unión; en las adiciones trans, el nucleófilo, a continuación ataca al ion carbonio más estable. A continuación se resume una serie de reacciones de adición y eliminación a un ciclohexano con impedimento estérico y con control estereoquímico de las mismas.
Reacciones de reducción.
Hidrogenación catalítica.
Es la adición de H₂ a los alquenos en presencia de catalizadores. Los más comunes son metales de transición, particularmente: Pt, Pd, Rh, Ni y Cu finamente divididos.
Excepto para olefinas estéricamente impedidas, esta reacción procede en forma muy rápida y ofrece buenos rendimientos. Los átomos de la superficie de un cristal de Ni, por ejemplo, tendrán seguramente valencias libres dirigidas hacia afuera del cristal y es significativo que tanto el etileno y el H₂ reaccionen exotérmica y reversiblemente con una supeficie libre del Ni.
En el caso del etileno, este ataque debe implicar los electrones π, puesto que la adsorción es imposible con el etano. Como la molécula de H₂ no dispone de electrones π, la adsorción observada debe provocar un debilitamiento muy considerable entre los átomos de hidrógeno.
El compuesto con múltiples enlaces interactúa con la superficie del catalizador formando intermediarios en los cuales la molécula orgánica está adsorbida fuertemente. Para alquenos simples se indican a continuación tres tipos de intermediarios posibles implicados en el proceso de hidrogenación.
Como se muestra en A, vemos el estado inicial en el que las moléculas de H₂ y la olefina se encuentran adsorbidos a la superficie del catalizador.

Como observamos en B, puede producirse la adición de uno de los hidrógenos conduciendo al resultado de una especie monoadsorbida unida a la superficie del metal por uniones semejantes a una unión π.

Este intermediario puede entonces reaccionar con el H₂ adsorbido y obtendremos el compuesto saturado. No debemos considerar uniforme a la superficie del metal. En ella, aparentemente hay sitios que muestran preferencia para tipos específicos de interacción con moléculas orgánicas.
También haremos notar que la superficie del catalizador no es regular, produciéndose interacciones estéricas variables con la molécula adsorbida.
En la mayoría de los casos, ambos átomos de H se adicionan del mismo lado del sustrato (adición syn). Esto puede ser el resultado de una adición prácticamente simultánea de ambos átomos de H. Si la hidrogenación ocurre en dos pasos, el intermediario debe permanecer unido a la superficie del metal para que no ocurra una inversión de la configuración por simple rotación sobre la unión C-C.

La adsorción sobre la superficie del catalizador se produce normalmente por el lado menos impedido de la molécula. Algunos ejemplos de dicha situación son los siguientes:


La mayoría de las reducciones catalíticas son llevadas a cabo en sistemas en los cuales el H₂ gaseoso es introducido desde cilindros comerciales de alta presión. Sin embargo, hay técnicas disponibles para generar H₂ químicamente (NaBH₄ + H⁺)
Estas técnicas se usan especialmente cuando se desea realizar hidrogenaciones a baja temperatura.
La hidrogenación catalítica de alquinos para obtener alquenos debe realizarse con catalizadores parcialmente envenenados, por ejemplo, H₂ sobre Pd/C en presencia de BaSO₄. En estos casos, la hidrogenación parcial también resulta ser syn.
Reducción química.
1) Hidroboración.
La adición de diboranos y boranos sustituidos a dobles enlaces C-C (C=C) es un importante método de síntesis de alcoholes. La reacción, llamada hidroboración, implica la adición en un único paso y procede rápidamente hasta que todas las uniones B-H hayan reaccionado generando trialquilboranos, salvo en el caso de olefinas estéricamente impedidas. El diborano puede ser generado "in situ" a partir de NaBH₄ y F₃B:
3 NaBH₄ + BF₃ ⟶ 2 B₂H₆ + 3 F₄BNa
CH₃-CH₂-CH₂-CH=CH₂ + B₂H₆ ⟶ (CH₃-CH₂-CH₂-CH₂-CH₂)₃B
Se ha observado que la adición de la unión B-H es altamente selectiva con respecto a la orientación y a la estereoquímica de la adición. El boro es menos electronegativo que el hidrógeno. Cualquier carga positiva que se desarrolle en el estado de transición, es siempre mejor acomodada por el extremo más sustituido de la múltiple unión.

La regla de Markovnikov se cumple para la adición, pero hay que tener en cuenta que el hidrógeno no es la posición más positiva en el reactivo atacante.
La reacción de hidroboración es también predecible con respecto a la estereoquímica de la adición. La adición es syn y sucede a través de un estado de transición de cuatro centros con la formación simultánea de las uniones C-B y C-H acompañada por la ruptura concertada de la unión B-H.

Desde el punto de vista de los orbitales moleculares, la reacción de adición se debe a la interacción del orbital π de la olefina con el orbital p vacío del boro trivalente. Los organoboranos son importantes debido a las otras reacciones que ellos pueden sufrir.
El átomo de boro puede ser reemplazado por OH, aminas y halógenos, la reacción más ampliamente usada de los organoboranos es la oxidación a alcoholes. El reactivo frecuentemente utilizado es el peróxido de hidrógeno en solución acuosa alcalina. El mecanismo se indica a continuación:

Agentes oxidantes más vigorosos producen el reemplazo del boro y la oxidación del átomo de carbono sustituido, permitiendo la síntesis de cetonas.

Otra posible reacción para los trialquilboranos es el tratamiento con ácido acético obteniéndose el alcano correspondiente:

2) Hidroxilación
La configuración del producto formado por hidroxilación de un doble enlace depende de la naturaleza del agente hidroxilante y de las condiciones en que se realiza la misma.
a) Hidroxilación cis
Tanto el OsO₄ como el KMnO₄ atacan rápidamente a los alquenos. En ambos casos se explica fácilmente la adición cis suponiendo que se forma un compuesto organometálico cíclico intermediario.
El KMnO₄ debe ser usado en condiciones muy controladas ya que produce generalmente una posterior oxidación del glicol, con clivaje de la olefina, para dar compuestos con mayor grado de oxidación.
El OsO₄ minimiza el problema de la sobreoxidación, pero tiene el inconveniente de ser muy tóxico y caro. El glicol es liberado del éster ósmico por reactivos como el ion bisulfito o el SH₂.
La hidroxilación con H₂O₂ catalizada por OsO₄ en butanol terciario es una adición cis; si la reacción se cataliza con dióxido de selenio en butanol terciario o acetona, la adición es trans.

b) Hidroxilación trans
Se da con perácidos. El primer paso es el ataque del perácido para formar un epóxido. Los más usados son: peroxiacético, peroxibenzoico, m-cloroperoxibenzoico, peroxitrifluoracético.
En el proceso de epoxidación no están involucrados intermediarios iónicos. La velocidad de la reacción no está relacionada con la polaridad de los solventes. Se produce una adición syn con retención de la estereoquímica de los sustituyentes de la olefina. Se cree que es un proceso concertado. El estado de transición puede representarse de la siguiente forma:

La reactividad de la olefina se incrementa con sustituyentes dadores de electrones y la de los perácidos con atractores de electrones.
La estereoquímica de la epoxidación con ácidos peroxicarboxílicos ha sido bien estudiada. El ataque y la adición de oxígeno ocurre preferentemente por el lado menos impedido de la molécula. En las moléculas donde hay dos modos potenciales de aproximación se obtienen mezclas.

La epoxidación es seguida de apertura nucleofílica o solvolítica del epóxido.
En sistemas cíclicos la apertura del anillo ocurre dando dioles diaxiales.

3) Adición a carbenos
Un número importante de reacciones involucra, como intermediarios, el uso de compuestos del carbono divalente altamente reactivo (carbeno). Estas especies son eléctricamente neutras, están unidas sólo a dos sustituyentes y poseen dos electrones no apareados. La más útil de las reacciones de carbenos es la adición a una doble unión para dar un ciclopropano. Esta reacción es estereoespecífica formando cada isómero geométrico el producto de adición cis.

4) Ozonólisis
La ruptura de alquenos con ozono es un procedimiento degradativo muy útil. Este reactivo rompe las uniones C=C con la formación de un peróxido cíclico conocido como ozónido. La posterior descomposición del ozónido produce grupos carbonilo sobre los carbonos unidos inicialmente por la doble unión.
El carácter electrofílico del ozono deriva del hecho de que cada uno de los tres átomos de oxígeno tiene una considerable afinidad por los electrones.

El mecanismo de la formación del ozónido implica un número de diferentes estados partiendo de una cis adición de ozono al alqueno.

5) Reacción de Diels-Alder
La adición térmica y concertada de un alqueno (dienófilo) a un dieno es altamente estereoespecífica. Esta reacción juega un papel importante en síntesis orgánica, ya que consiste en la unión de dos esqueletos carbonados en forma directa y estereoespecífica. La estereoquímica del dieno y del dienófilo es retenida en el proceso de cicloadición y siempre se forma un anillos de seis miembros por adición 1,4-cis. El dienófilo es activado por sustituyentes que atraen electrones (-COOH, -COH, -NO₂, -CN y -SO₂) mientras que los dienos se ven activados por sustituyentes dadores de electrones.

El estado de transición para la adición necesita que el dieno adopte la conformación cis. El dieno y el dienófilo se aproximan en planos paralelos estableciéndose en ese momento una serie de interacciones de unión entre el carbono 1 y el 4 del dieno y los átomos de carbono de la doble unión del dienófilo.
Existe un rasgo adicional en lo que se refiere a la estereoquímica de la reacción, y que especialmente se pone de manifiesto en la adición de dienófilos a dienos cíclicos. Esto implica la orientación relativa del dieno y del dienófilo en el estado de transición, que puede conducir a dos productos, el endo y el exo.
Estos términos indican que los sustituyentes están dirigidos hacia el interior o el exterior de la forma bote del ciclohexeno. En la práctica, el isómero endo es el que se forma, especialmente cuando los sustituyentes del dieno son grupos insaturados, aunque este isómero es el termodinámicamente menos estable (estéricamente más congestionado). La razón de este resultado se debe a una combinación de atracciones dipolares y de van der Waals, así como también a interacciones de orbitales que implican a los sustituyentes del dienófilo dando mayor estabilidad al estado de transición que conduce al isómero endo.

6) Adiciones polares de los halógenos y haloácidos.
Los halógenos y los haloácidos se adicionan rápidamente a los alquenos; en el primer caso el electrófilo es X⁺ (X₂ activado como X⁻ y X⁺) y en el último es el H⁺. Se conocen ejemplos de ambas adiciones, generalmente proceden en corto tiempo a temperatura ambiente y son predominantemente trans. En el caso de los ácidos hipohalogenados, XOH, el electrófilo activo también es X⁺. En la adición común de los ácidos halogenados (HCl y HBr) el protón es el electrófilo formándose un carbocatión que luego es atacado desde el lado opuesto por el anión.

En la reacción del bromo con alquenos, el primer paso es la tansferencia de un catión X⁺ al hidrocarburo y formación de un ion bromonio. El ataque del bromuro ocurre por el lado opuesto al ion bromonio, dando la trans adición de Br₂.

Reacciones de eliminación.
Es un hecho conocido que para que se produzca una reacción de eliminación, los átomos intervinientes deben ser coplanares y estar en posición trans uno respecto del otro.

Si esto no ocurre, la eliminación no se produce o se hace con mucha dificultad.
Veamos el caso del tosilato de 4-terbutil-ciclohexilo (I). Este compuesto debido al volumen del grupo terbutilo se presenta siempre con dicho grupo en ecuatorial pudiendo estar el grupo tosilato en axial o ecuatorial.

Podemos ver que en la forma cis el grupo TsO⁻ puede eliminarse ya sea con los átomos de hidrógeno axiales unidos a C₂ o C₆, dando el 4-terbutil-ciclohexeno, mientras que, en la forma trans, no ocurre eliminación ya que la molécula no posee en 2 o 6 átomos de hidrógeno coplanares y trans respecto al grupo tosilo.
En cambio, si los sustituyentes de la molécula no impiden el pasaje de una forma silla a la otra forma silla, la reacción puede ocurrir en ambos casos, pero con distinta velocidad, por ejemplo:

Otro caso lo tenemos en la reacción de deshidrohalogenación del cloruro de neomentilo y de mentilo

En el primer caso, la eliminación puede ocurrir con los hidrógenos axiales de C₁ y C₃, como así ocurre, pero la reacción obedece la regla de Saytzeff, predominando el 1-menteno sobre el 2-menteno.
En el segundo caso, para que se produzca la eliminación, la molécula debe pasar a la otra forma silla, que posee hidrógeno axial únicamente (a los efectos de la eliminación) en C₃ dando un único producto, el 2-menteno, sin seguir la regla de Saytzeff.
La estereoquímica de las reacciones también puede estar relacionada con la configuración de los sustituyentes, como por ejemplo en la formación de epóxidos por deshidrohalogenación de halohidrinas.
Un ejemplo de esto lo tenemos en el 3-α-clorocolestano-2-β-ol y el 2-β-clorocolestano-3-α-ol, que dan dos epóxidos isómeros:

Regla de Bredt: Las reacciones de eliminación en ciertos compuestos orgánicos está también regulada por determinados factores que están comprendidos en la regla de Bredt. Esta regla tiene validez en compuestos bicíclicos rígidos

La misma dice que: "en compuestos bicíclicos rígidos no es posible formar un doble enlace en el que participe un átomo cabeza de puente". Tal es el caso de la reacción anterior en que el C₄ formaría parte del doble enlace; en este caso la reacción no ocurre.
Las reacciones de eliminación para obtener el alqueno menos sustituido de los posibles (eliminación de Hoffman) puede ejemplificarse de la siguiente forma:

Debe notarse que el grupo amino debe estar en anti-diaxial respecto del -H con el que la eliminación se produce.
Reacciones de sustitución
En las reacciones de sustitución en sistemas cíclicos debemos tratar por separado los casos en que el sustituyente ataca al átomo que no está unido directamente al anillo (sustituyentes exocíclicos) de aquellos en que está unido a un carbono del anillo, pues los factores que intervienen son distintos. En el caso de las reacciones en posiciones exocíclicas, los ejemplos más estudiados son los de esterificación de alcoholes y saponificación de ésteres. En general, podemos decir que reaccionan más fácilmente los sustituyentes en ecuatorial. Por ejemplo:
 En este caso, el isómero trans (diecuatorial) se hidroliza 20 veces más rápido que el cis (ecuatorial-axial). Esta circunstancia puede atribuirse a efectos estéricos ya que el ataque a un sustituyente ecuatorial es más fácil por estar más alejado de los átomos vecinos.
En este caso, el isómero trans (diecuatorial) se hidroliza 20 veces más rápido que el cis (ecuatorial-axial). Esta circunstancia puede atribuirse a efectos estéricos ya que el ataque a un sustituyente ecuatorial es más fácil por estar más alejado de los átomos vecinos.
Sin embargo, en algunos sistemas rígidos, esta regla puede fallar, como en el caso del 3-β-acetoxicolestano-5-α-ol y su epímero 3α,


Sin embargo, en algunos sistemas rígidos, esta regla puede fallar, como en el caso del 3-β-acetoxicolestano-5-α-ol y su epímero 3α,

(en donde el grupo acetoxi es cis-axial con respecto al -OH axial) pues el epímero 3α se hidroliza con mayor facilidad que el 3β a pesar de que esté en ecuatorial. Esta discrepancia puede justificarse teniendo en cuenta que puede formarse puente de hidrógeno entre ambos grupos funcionales, lo cual facilitaría la solvólisis del éster.

En los sistemas cíclicos no-rígidos, la facilidad del ataque dependerá de la mayor o menor posibilidad que puede tener el grupo funcional en adoptar una posición favorable para reaccionar (y la presencia de otros sustituyentes).
Veamos ahora las reacciones de sustitución en las posiciones nucleares. La reactividad de los sustituyentes dependerá del mecanismo de la reacción, según sea SN₁ o SN₂.
En las reacciones del tipo SN₁ el factor predominante es la formación del ion carbonio, y a partir de éste, la entrada del nucleófilo estará orientada por la presencia de sustituyentes, pudiendo haber retención de la configuración o racemización o inversión, ya que el ataque se producirá por la parte menos impedida estéricamente.
En cambio, en las reacciones SN₂, el factor predominante es el estado de transición y la reacción ocurrirá más o menos fácilmente según la relación entre las energías del estado de transición y el estado fundamental de la molécula.
El estado de transición en el desplazamiento de un grupo ecuatorial, que presupone un ataque por la retaguardia, tiene un contenido energético mayor que el estado de transición para el desplazamiento de un grupo axial, que se produce también por la retaguardia para quedar por último, el grupo entrante en posición ecuatorial. En menor grado influye el impedimento que pueden presentar el grupo saliente los sustituyentes presentes en la molécula (en posición cis-axial, debido a un efecto de compresión) pero generalmente es compensado por el menor impedimento que se le presenta al reactivo atacante por la posición ecuatorial. En estas reacciones se produce inversión de la configuración.

Esta circunstancia hace que un sustituyente en posición axial sea más fácilmente sustituido que uno en ecuatorial.
Oxidación de alcoholes cíclicos.
La reacción general de oxidación con ácido crómico puede simplificarse así:

El cromo (IV) producido en el paso inicial no es estable y puede contribuir a continuar la oxidación, lo cual explica a su vez la formación de cromo (III) según:











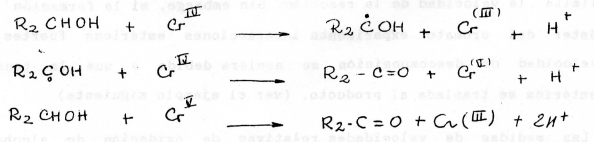
Otra forma en que se encuentra explicando el proceso es la siguiente:

En la oxidación de alcoholes cíclicos vale lo visto para las reacciones SN₂ pues el ataque se produce sobre la unión C-H, y por lo tanto un alcohol axial será más fácilmente oxidado que uno ecuatorial.
Un ejemplo de esto lo tenemos en el 4-terbutilciclohexanol en el que el isómero cis se oxida 3 veces más rápido que el trans.
El mecanismo es:

La velocidad de reacción se ve influida por la estabilidad del éster del cromato formado como producto intermedio. Si éste experimenta interacciones estéricas fuertes, lo que implica disminución de la estabilidad, la velocidad de descomposición se acelera.
Con alcoholes ecuatoriales, el equilibrio inicial para formar el éster de cromato es rápido, la descomposición de dicho éster es lo que limita la velocidad de reacción. Sin embargo, si la formación del éster del cromato experimenta interacciones estéricas fuertes, la velocidad de descomposición se acelera debido a que la tensión estérica se traslada al producto (ver el ejemplo siguiente).
Las medidas de velocidades relativas de oxidación de alcoholes epímeros con ácido crómico a sus cetonas correspondientes han sido particularmente útiles para hacer asignaciones estereoquímicas ya que debido a la ausencia de reacciones secundarias importantes se oxida más rápidamente el alcohol con el grupo hidroxilo más impedido.

Reducción de cetonas cíclicas.
a) Reducción catalítica
La presencia de ácidos y bases en las mezclas reactivas empleadas para la hidrogenación catalítica altera, en muchos casos, el porcentaje y el curso estérico de muchas reducciones. Así, por ejemplo, para las reducciones de los derivados de la ciclohexanona, la hidrogenación en medio neutro o básico produce predominantemente alcoholes de tipo ecuatorial, mientras que los de mayor contenido energético (los axiales), son los productos principales de la reducción en medio ácido. Este postulado rige para cetonas cíclicas rígidas y se conoce con el nombre de Regla de Auwers-Skita. Sin embargo, esta regla no es absoluta ya en medio -OH, o neutro o H⁺, siempre se producirá algo del otro isómero.

b) Reducciones con hidruros metálicos
La hidrogenación catalítica presenta un problema, que consiste en la imposibilidad de reducir selectivamente las funciones carbonilo de las cetonas, ésteres y amidas en presencia de dobles enlaces C=C, lo que ha conducido al empleo de hidruros metálicos complejos. Las sales asequibles comercialmente son, enunciadas en orden de reactividad decreciente: H₄LiAL, BH₄Na, etc.

La reacción del carbonilo con el hidruro, implica la transferencia del ion hidruro al átomo de carbono sp². Este paso, en un solvente no prótico, viene seguido por formación de un anión alcoxiborohidruro, mientras que en solventes hidroxílicos, se puede producir un intercambio con el disolvente. Una secuencia comparable a la reacción del borohidruro en un disolvente no protónico, se aplica a las reducciones con H₄LiAl

Aunque se han estudiado las reducciones de un gran número de cetonas cíclicas con hidruros metálicos, es difícil predecir el resultado estereoquímico de dichas reacciones.
Se han encontrado dos factores que pueden determinar el curso de la reacción de reducción con H₄LiAl: el impedimento estérico a la aproximación del hidruro metálico a la función carbonilo, llamado control de aproximación estérico, y la estabilidad del producto final, llamado control de evolución del producto. Si se usan las estructuras 1 y 2 para representar los estados de transición, para las dos direcciones posibles de reducción, de derivados de la ciclohexanona conformacionalmente rígida, se puede ver que la interferencia estérica entre los grupos R₁ y R₂ hacia arriba y la aproximación por dicho lado del hidruro metálico (control de evolución del producto) se opondrán a la formación del alcohol ecuatorial (el más estable).

Por otro lado, la aproximación del H₄Al⁻ de modo que la interferencia estérica entre él y los grupos R₁ y R₂ sea mínima en 2 (control de aproximación estérica) llevarán a la formación del alcohol axial 4. Por lo tanto, si la aproximación a un lado de la función carbonilo está mucho más impedida que la aproximación por el otro, como en la reducción del alcanfor (5), se obtiene una mezcla de alcoholes en la cual predomina el alcohol que se produce por el control de aproximación estérica independientemente de la estabilidad del producto final.

Se ilustra a continuación la formación primaria de un alcohol ecuatorial (6) al reducir la cetona (7)

En este caso, donde no existe el impedimento para el acercamiento del reactivo, es decir, donde el control de aproximación estérica no es un factor decisivo, el producto que se obtiene es el favorecido por su mayor estabilidad, es decir, el alcohol ecuatorial.
Descarga: Análisis conformacional y estereoquímica de las reacciones
No hay comentarios:
Publicar un comentario